Hello.
I have a question for you.
Are you using Cartesian coordinates? If you answer is yes, you have to be careful the Cartesian coordinates is actived instead of the internal ones. By default, the jobbsse script activate both internal coordinates and redundant coordinates. This option don't have problems for working with jobbsse script because this one only makes a serial of singlepoints calculations.
In order to do a geometry optimization you have two options:
-Make a jobbsse calculation and after you have to take the imputs, which was built in n°_ghosts folders (e.g 1_ghost). Here, you have to perform a geometry optimization by using jobex script (you must remember to check what kind of coordinates are activated in control file).
-The another option is you have to build the input file manually. You can do this by editing the control file. When you want to assign the group of atoms like ghost atoms, you must add the line charge = 0.00000 below of the basis line.
I adjunct the next example:
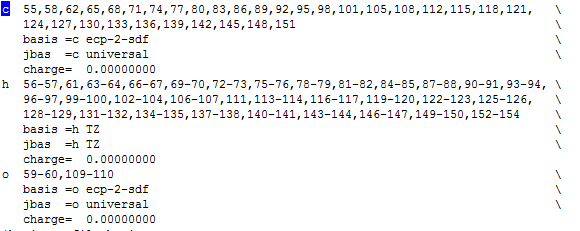
For my own experience, I advice you don't do a geometry optimization by using ghost basis. I don't recommended that because these kind of calculation may have convergence problems (I lived that hehehehe).
I recommend you perform a normal jobbsse calculation and after you take the corrected energy value of complete system. you may optimize the monomers molecules without BSSE correction and you may compare the energies with the energy corrected.
I hope to help you with this post
Cheers

